KAIST 화학과 김우연 교수팀이 물리법칙을 스스로 이해해 분자 구조를 예측하고 설계하는 인공지능(AI) 모델 ‘리만 확산모델(R-DM)’을 개발, 신약이나 신소재 개발 속도를 획기적으로 높이는 발판을 마련했다.
스마트폰 배터리 수명이나 난치병 치료제 성능은 재료를 구성하는 원자들이 얼마나 안정적으로 결합하느냐에 달려 있다.
기존 AI는 단순히 분자 모양의 좌표를 흉내 내는 수준으로, 실제 화학 반응에서 중요한 결합 길이와 에너지 변화를 정밀하게 반영하지 못하는 한계가 있었다.
연구팀은 이런 한계를 극복하기 위해 리만 기하학을 AI에 접목했다.
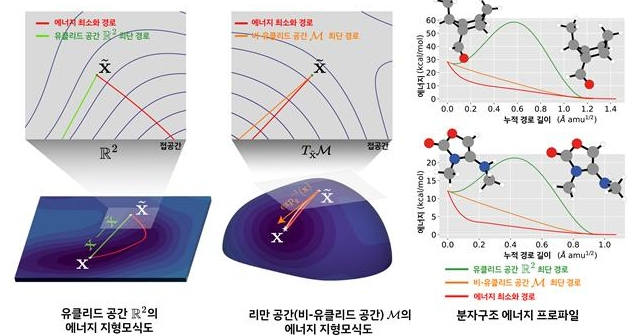
연구팀은 분자 구조를 에너지가 높으면 언덕, 낮으면 골짜기로 표현한 지도로 나타내고, AI가 에너지 지형에 따라 구부러진 공간 위에서 가장 안정적인 상태인 골짜기를 스스로 찾아 이동하도록 설계했다.
이는 ‘물질은 에너지가 가장 낮은 상태를 선호한다’는 화학 기본 원리를 AI가 수학적으로 학습하도록 만든 결과다.
기존 모델이 평평한 공간에서 원자 좌표만 이동시켰다면, 이번에 개발한 기술은 실제 에너지 흐름을 따라 가장 안정적인 구조를 완성한다.
실험 결과 리만 확산모델은 기존 AI보다 20배 이상 높은 정확도를 보였고, 예측 오차는 정밀한 양자역학 계산과 거의 차이가 없는 ‘화학 정확도(1 kcal/mol 이하)’ 수준을 달성했다.
이 기술은 고비용의 양자역학 시뮬레이션을 대체하거나 보완할 수 있는 AI 시뮬레이터로 활용할 수 있을 전망이다.
특히 신약 개발은 물론 차세대 배터리 소재, 고성능 촉매 설계 등 정밀한 구조 탐색이 필요한 산업 전반의 연구개발 속도를 획기적으로 높일 것으로 기대된다.
아울러 실험이 어렵거나 위험한 화학 사고, 유해물질 확산경로 예측에도 적용할 수 있어 환경과 안전 분야에서도 활용 가치가 크다.
김 교수는 “AI가 화학 기본원리를 이해하고 분자 안정성을 스스로 판단한 첫 사례”라며 “신소재 개발 방식을 근본적으로 바꿀 수 있는 기술”이라고 설명했다.
한편, 이번 연구는 KISTI 슈퍼컴퓨팅센터 우제헌 박사와 KAIST 혁신신약연구단 김성환 박사가 공동 제1저자로 수행했고, 연구결과는 지난달 2일 국제학술지 ‘네이처 컴퓨테이셔널 사이언스(Nature Computational Science)’에 게재됐다.
(논문명: Riemannian Denoising Model for Molecular Structure Optimization with Chemical Accuracy, DOI: 10.1038/s43588-025-00919-1)